

一种N掺杂γ-Fe2O3电催化剂
电子说
描述
最新成果介绍
电催化CO2还原(CO2RR)为CO2的高效利用提供了途径。然而,到目前为止,Cu仍然是CO2RR生成C2+产物的主要活性组分。在非Cu基CO2RR电催化剂上生成C2+产物的报道较少,且效率较低。中科院化学研究所Zhang Pei、韩布兴院士等人报道了一种N掺杂γ-Fe2O3电催化剂(xFe2O3-N@CN),其在H型电解槽中可催化CO2生成C2H6。在-2.0 V vs Ag/Ag+时,电流密度为32 mA cm-2,C2H6的法拉第效率达到42%。值得注意的是,这是首次报道在Fe基催化剂上来实现CO2还原生成C2H6。 结果表明,催化剂中具有富含氧空位的FeO1.5-nNn位点,有利于*COOH中间体的稳定。两个相邻表面Fe原子的暴露,有利于降低FeO1.5-nNn位点上C-C耦合的能垒,从而促进C2H6的生成。
相关工作以《Efficient Electrocatalytic Reduction of CO2 to Ethane over Nitrogen-Doped Fe2O3》为题在《Journal of the American Chemical Society》上发表论文。
图文介绍
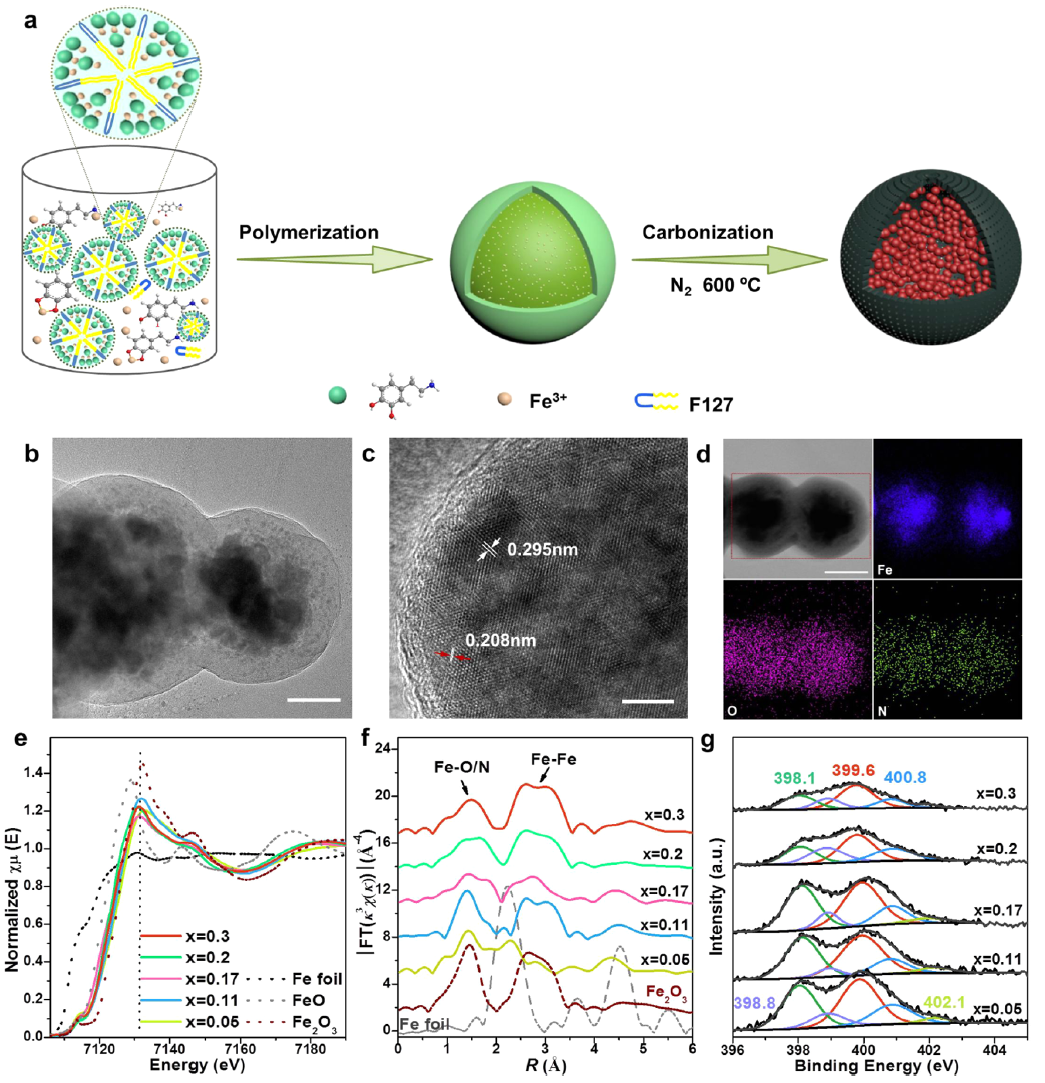
图1xFe2O3-N@CN的制备和结构表征以F127为模板,在含Fe3+的缓冲溶液中,多巴胺(DA)发生原位聚合。将制备好的纳米颗粒在N2气氛下进行煅烧后,得到了带有CN壳层的N掺杂Fe2O3,如图1a所示。根据ICP-OES结果,将不同Fe含量的样品记为xFe2O3-N@CN,下标x表示碳化后所有元素中Fe的摩尔分数。以0.3Fe2O3-N@CN为例,TEM图像显示催化剂具有核-壳结构,壳层厚度约30 nm。 所制备的xFe2O3-N@CN的Fe的K边XANES曲线和一阶导数表明,近边吸收曲线介于Fe箔和Fe2O3之间,表明Fe处于Fe3+氧化态。在FT-EXAFS曲线中,在1.4~1.7 Å处出现了一个宽的峰,这可能是Fe-O/N配位的结果,而在2~3 Å附近的峰对应Fe-Fe配位。定量EXAFS拟合分析表明xFe2O3-N@CN中Fe-O/N的配位数均低于Fe2O3的配位数,表明Fe-O/N具有不饱和配位构型。
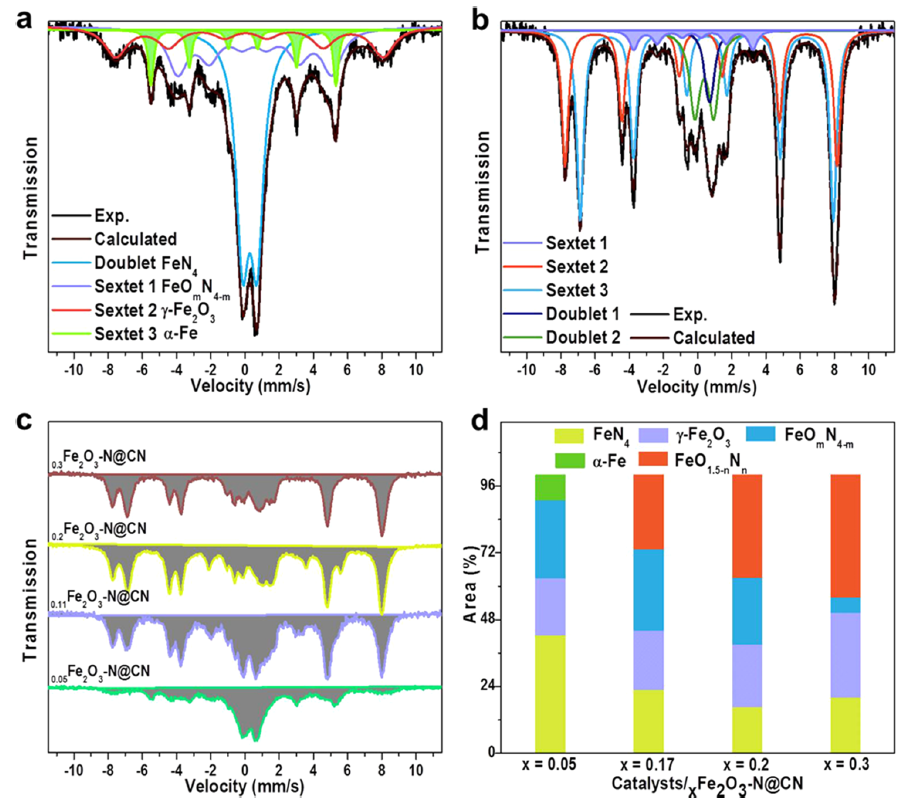
图2 57Fe Mössbauer光谱表征为了更好地识别催化剂中Fe物种的局部配位,进行了57Fe Mössbauer光谱测量来区分铁基催化剂的详细结构。结果显示,配位环境随Fe含量的变化而变化,Fe含量高的催化剂中存在更多富氧的FeO1.5-nNn位点和氧缺陷。
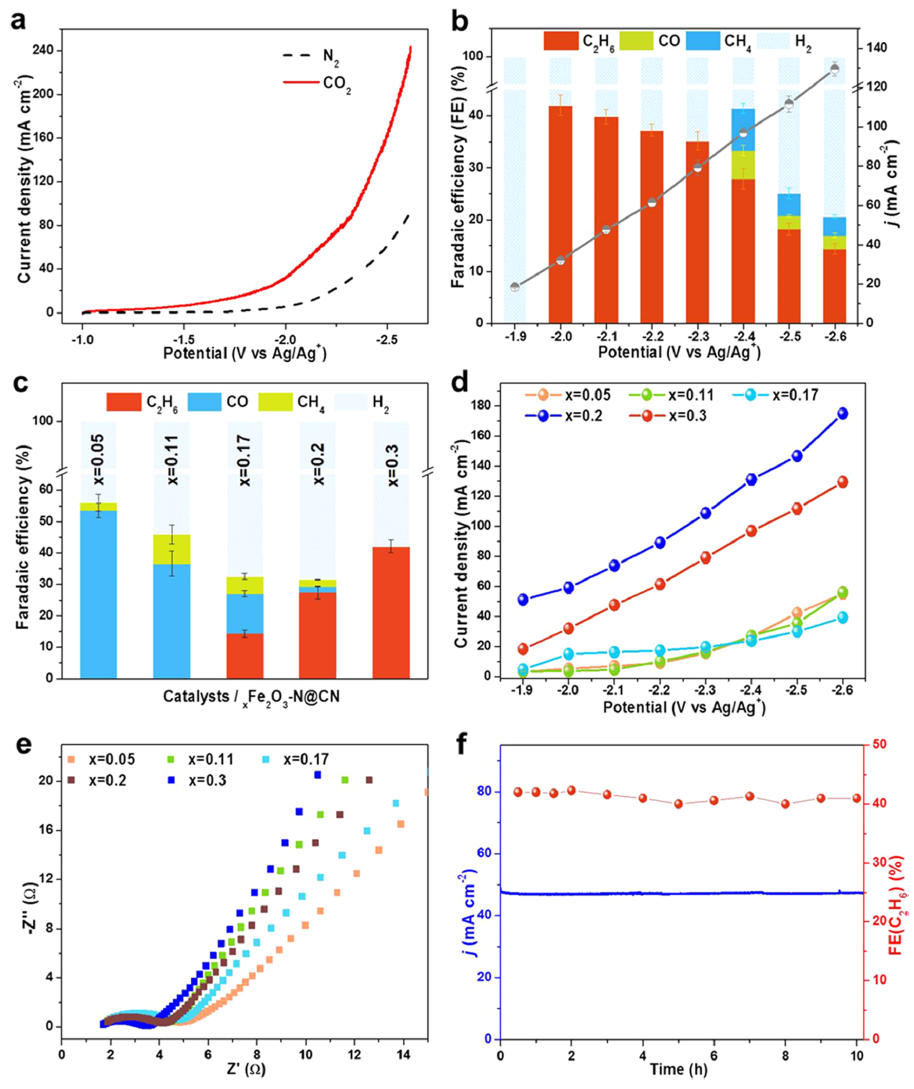
图3 在[Bmim]PF6/MeCN/H2O中进行CO2RR在离子液体(IL)基电解质的三电极体系中,评价了Fe含量最高的0.3Fe2O3-N@CN的催化CO2RR性能。极化曲线显示,与N2饱和的电解质相比,在CO2饱和的电解质中0.3Fe2O3-N@CN上产生了更高的电流密度,表明0.3Fe2O3-N@CN 具有CO2RR活性。在-1.9~-2.6 V vs Ag/Ag+下测定了电流密度与电解产物的法拉第效率。其中,在-2.0 V vs Ag/Ag+下,电流密度为32 mA cm-2,C2H6的法拉第效率达到42%,且仅有唯一的副产物H2。同时,在xFe2O3-N@CN 中,FEC2H6和电流密度随Fe含量的增加而增大。Fe含量和由此产生的Fe-O-N配位是制备的xFe2O3-N@CN的主要差异,这决定了CO2RR中生成C1或C2产物的选择性。
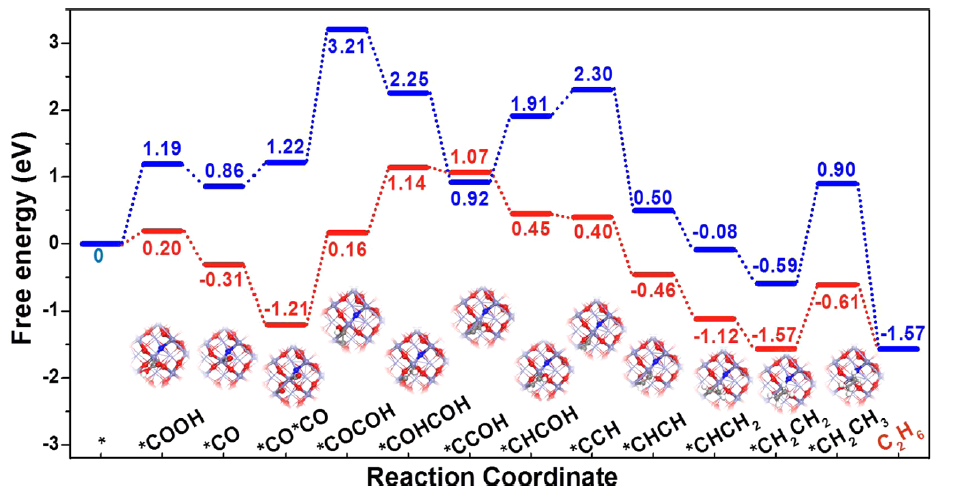
图4 DFT计算通过DFT计算进一步揭示了FeO1.5-nNn活性物种的CO2RR性能。首先建立了N掺杂Fe2O3(FeO1.5-nNn)和原始Fe2O3两种催化剂模型进行计算。图4显示了CO2还原成C2H6的自由能变化,其中红线为N掺杂Fe2O3催化剂,蓝线为Fe2O3催化剂。可以看出,在CO2的第一步加氢过程,N掺杂Fe2O3催化剂上自由能变化仅为0.2 eV,远低于Fe2O3催化剂。后续,在速率限制步骤C-C耦合阶段,N掺杂Fe2O3催化剂上反应能垒为1.37 eV,低于Fe2O3催化剂的1.99 eV。 DFT计算揭示,xFe2O3-N@CN生成C2H6的高活性是由于*COOH中间体在FeO1.5-nNn位点上的吸附和稳定,并暴露在相邻的Fe表面,导致C-C耦合过程的能垒降低。这项工作是首次在Fe基催化剂上进行电催化CO2转化为C2产物的成功案例。本研究为新型铁基催化剂的设计提供了一种策略,该催化剂具有可调的局部配位和电子结构,可将CO2转化为CO2RR中的C2产物。
文献信息
Efficient Electrocatalytic Reduction of CO2 to Ethane over Nitrogen-Doped Fe2O3,Journal of the American Chemical Society,2022. https://pubs.acs.org/doi/10.1021/jacs.2c05373
审核编辑 :李倩
-
碱性醇类燃料电池新型催化剂的研究2011-03-11 0
-
燃料电池氧电极催化剂的研究2011-03-11 0
-
研究人员开发出一种新型催化剂材料 将推动氢能源发展2020-03-25 1207
-
高效电催化二氧化碳还原反应催化剂成功研制2020-03-30 4776
-
低结晶和异质结构AuPt-Ru@CNT像高效多功能电催化剂2022-05-31 445
-
调控锂盐消除催化剂表面凝胶化构筑高比能锂硫电池2022-07-13 1736
-
一种RhCu SAA催化剂的原理设计2022-07-22 522
-
Mo配位FeCoNiMo碳负载高熵电催化析氧催化剂图文解析2022-09-20 2121
-
析氢反应(HER)电催化剂在电解装置的广泛应用2022-09-28 9032
-
蜂窝状多孔结晶异质电催化剂实现高效的CO2吸附/活化2022-09-30 2537
-
碳纳米管桥接策略用于双功能氧电催化剂2022-11-11 1406
-
如何提高HEAs催化剂的催化活性和优选设计研究2022-12-14 1021
-
一种用于高效PEMFCs的紧密填充的混合Pt1.5Ni1-x/Ni-N-C电催化剂2022-12-26 1121
-
揭示卤素掺杂Sn基催化剂促进CO2电还原制甲酸盐原因2022-12-29 2514
-
依赖于尺寸和载体的Ptn/X-石墨烯催化剂(X = C、B、N)减弱CO中毒2023-01-11 1002
全部0条评论

快来发表一下你的评论吧 !
