

调节可充电锂电池中LiNi0.5Mn1.5O4实现锰镍氧化还原
描述
文章一:调节可充电锂电池中LiNi0.5Mn1.5O4尖晶石正极动态电化学界面实现锰镍同时氧化还原
一、 全文概要
爆炸式增长的电动汽车市场需要高性价比的高能材料来制造可充电锂电池。基于多阳离子(Ni2+/Ni4+和Mn3+/Mn4+)氧化还原中心的富锰尖晶石氧化物LiNi0.5Mn1.5O4(LNMO)可存储容量大于200 mAh g−1。但由于Mn3+/Mn4+氧化还原的可逆性较差,其实际容量仅限于Ni2+/Ni4+氧化还原(135 mAh g−1)。这种不稳定性通常归因于Mn3+的Jahn-Teller扭曲及其歧化,从而导致严重的Mn溶解。
韩国科学技术研究院Jihyun Hong团队首次证明了Mn3+/Mn4+氧化还原在2.3-4.3 V范围内具有良好的可逆性。LNMO只在2.3-4.9 V的较宽电压范围内失去容量。结果表明,循环过程中会反复发生电位驱动的岩盐相形成和分解等电化学界面的动态演化过程。
界面演化导致电解液降解和表面钝化,阻碍了电荷转移反应的进行。进一步证明,通过电解液改性稳定界面,在使用多阳离子氧化还原的同时延长了LNMO的循环寿命,使LNMO在500次循环后仍能保持71.5%的容量。该动态氧化界面为实现锰的可逆氧化还原,开发富锰正极提供了新的思路。
二、 研究亮点
1、 利用表面敏感的结构探针和化学探针,重新探讨了镍和锰的多阳离子氧化还原反应时LNMO正极的破坏机理。
2、 证明了Mn3+/Mn4+氧化还原反应前所未有的高可逆性,推翻了传统的LNMO降解理论。将这种恶化归因于由电位变化引起的电化学界面的动态演化。
3、 证明了简单的电解质修饰(例如,EC排除)使LNMO在100次循环后具有91.0%的容量保持率,能量密度为715 Wh kg-1,功率密度为588 W kg-1,电压范围为2.3-4.9 V。
三、正文导读
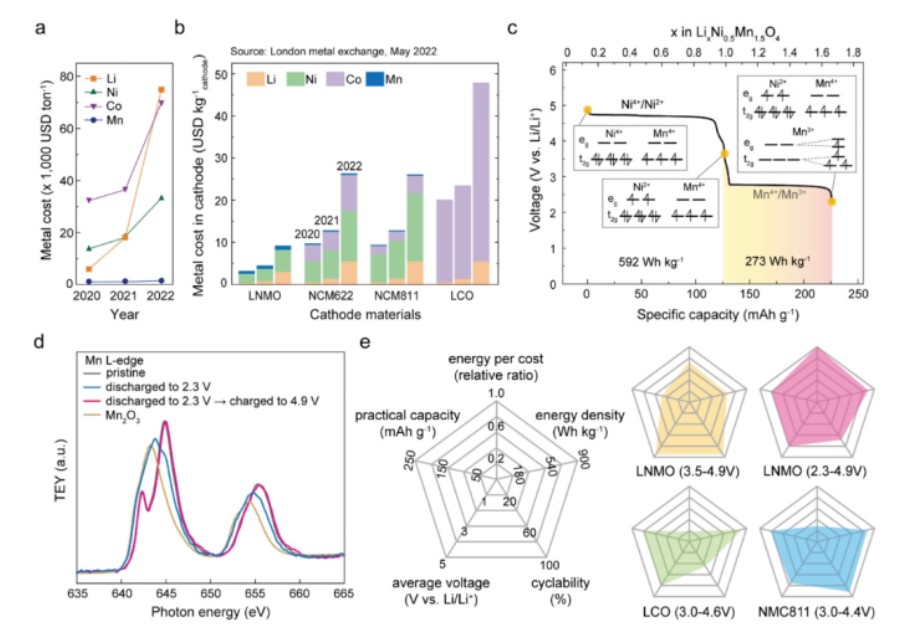
【图1】a)每年Li, Ni, Co, Mn的成本和b)每公斤正极活性材料的组成金属。c)在0.05 C下,在4.9-2.3 V电压下放电过程中,同时具有Ni和Mn氧化还原能力的LNMO的电压分布图和相应的Ni和Mn的电子构型。d) 0.05 C下LNMO的Mn L2,3边NEXAFS TEY光谱。e) LNMO、LCO和NMC811正极材料的蜘蛛图比较。为了进行公平的比较,LCO值是基于4.6 V充电截止的工作计算的。
(1)LiNi0.5Mn1.5O4的循环性的电压范围依赖性
为了检验同时使用锰和镍氧化还原偶的可行性,在0.2C下测试了LNMO半电池在三个不同电压范围下的循环性能:2.3-4.3 V(低电压范围,LVR)、3.5-4.9 V(高电压范围,UVR)和2.3-4.9 V(WVR),如图2a所示。在测试中,使用了平均尺寸为3.5 μm的商用LNMO颗粒。LNMO电池在LVR中经过100个循环后仍保留了初始容量的96.2%,其中Mn3+/Mn4+氧化还原反应导致了2.7V和4.1V的电压平台(图2b)。
在较长的循环过程中,过电位几乎没有增加,从微分容量与电压(dQ/dV)曲线可以观察到,库仑效率(CE)保持在99.2%以上。这一结果与传统主张的三价锰对电池循环寿命的有害影响相矛盾,因为它会产生Jahn-Teller畸变,然后是歧化。事实上,这一结果证实了在高压尖晶石中可逆利用Mn3+/Mn4+氧化还原反应达到超过850 Wh kg−1的高能量密度的可行性。
在使用Ni2+/Ni4+氧化还原的UVR中,即常规电压范围内,LNMO电池在100次循环后仍保持95.3%的初始容量(图2c)。这个容量保留值与以前报告中的值一致。然而,当在WVR中同时使用Ni2+/Ni4+和Mn3+/Mn4+氧化还原偶联时,LNMO电池的容量迅速下降,100次循环后仅保留56.1%(图2d)。dQ/dV曲线表明过电位显著增加,反映了在WVR循环过程中电池阻抗的增长。
同样的现象,即循环性的电压-范围依赖性,在1 C的较高电流密度下观察到。LNMO电池在WVR中经过300个周期后损失了53.8%的初始容量,而在UVR和LVR中分别损失了8.1%和8.5%。尽管WVR在1 C时的可逆容量较低,但LNMO正极的快速降解是无法避免的。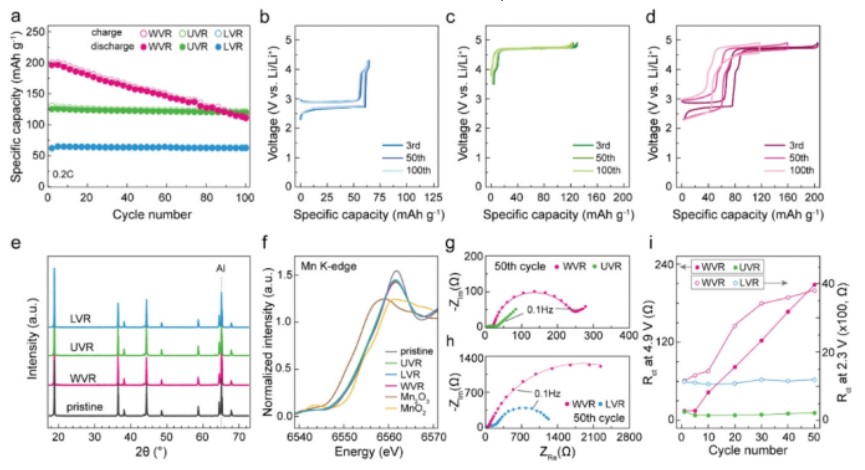
【图2】a)LNMO在0.2 C下LVR、UVR和WVR中的循环性能。LNMO在0.2 C下LVR、c)UVR和d)0.2 C下WVR下的第3、50和100个电压曲线。e)原始LNMO电极和LNMO电极在1 C下LVR、UVR和WVR内循环100次后的XRD曲线和f)Mn K边XANES光谱。LNMO半电池在0.2 C g)的WVR和UVR测量在4.9 V和h)的WVR和LVR测量在2.3 V的50个周期后的EIS曲线。i)LNMO在WVR、UVR和LVR中循环时电荷转移电阻(Rct)的演变。
LNMO在LVR和WVR中循环寿命的鲜明对比表明,仅仅使用Mn3+/Mn4+氧化还原反应并不一定会导致容量损失和电阻增加。
因此,破译LNMO在LVR和WVR中的不同降解机制,对于实现多阳离子氧化还原反应在富锰正极材料中的完全应用至关重要。首先用X射线衍射(XRD)和基于同步加速器的X射线吸收光谱研究了LNMO在三个电压范围内分别经过100次循环后的体积特性。所有样品在测量前放电到3.5 V,以匹配Li化学计量和TM氧化态。
从图2e的XRD谱图可以看出,在任何电压范围内,LNMO尖晶石的晶体结构几乎没有变化,没有形成任何二次相。全模式拟合结果显示,所有循环LNMO的晶格参数为8.17 Å(空间组:Fd3(-)m),与原始LNMO的晶格参数相同。(111)峰的全宽-半最大值保持相似,标准差小于5%,表明循环过程中体结构几乎没有变化。
在LVR和WVR中循环的LNMO正极发生立方(Fd3(-)m)和四方(I41/amd)相之间的可逆相变,并经历Mn3+的Jahn-Teller畸变。但在3.5 V条件下,经过长时间循环后,它们的体结构与LNMO在UVR条件下循环后的体结构几乎没有区别,其中Mn几乎不发生氧化还原。
图2f中的Mn K边X射线吸收近边谱(XANES)仅捕捉到100次循环后放电至3.5V的LNMO之间Mn氧化态(+4)的微小差异。Nik边XANES在Ni氧化态的对比也可以忽略不。总体结果证实,无论容量损失多少,体积退化都可以忽略不计。
相反,电化学阻抗谱(EIS)分析显示,在不同电压范围内,电池电阻有显著差异。由于Nyquist图在3.5 V下的半圆特征不明确,比较了在4.9V下WVR和UVR中循环的电池和在2.3V下WVR和LVR中循环的电池的EIS光谱。如图2g-i所示,在50个WVR周期中,电池的电荷转移电阻(Rct)持续显著增加,而UVR和LVR周期电池的Rct值保持稳定。
Rct的增加主要是导致WVR过电位的增长,这是由于CEI上的副反应阻碍了电荷转移反应。在WVR中,第50个周期的Rct值在4.9 V和2.3 V时分别对应于第5个周期的14倍和3倍。在不同电位下观察到的差异暗示了CEI在特定电压区域演化的可能性。
(2)低压驱动LiNi0.5Mn1.5O4表面尖晶石-岩盐相变
为了研究电压范围-界面的相关性,使用高分辨率透射电子显微镜(HR-TEM)结合空间分辨电子能量损失谱(EELS)和软X射线吸收光谱彻底研究了循环LNMO表面的晶体学和化学结构。
在分析过程中,所有电极在1 C下循环100次后放电至3.5 V。尽管在三个不同电压范围内LNMO的所有选定区域衍射模式都显示为尖晶石相,与原始状态相同,但HR-TEM图像清楚地揭示了LNMO独特的表面结构(图3a-c)。在LVR中循环后,LNMO表面转变为厚度为7-10 nm的岩盐相,通过10×10 nm2面积的快速傅里叶变换(FFT)模式探测到(图3a)。
在岩盐层内部,原始尖晶石相保持完整。HR-TEM图像和FFT图中晶格条纹的平行方向表明,岩盐相与尖晶石相是相干的,共享氧格架。相干性是氧释放引起阳离子致密化的证据。从表面到本体间隔4 nm处收集到的Mn L边EELS光谱显示了距离表面8 nm处的二价锰阳离子。
相比之下,在UVR中循环后,LNMO表面保持尖晶石结构,这由FFT模式和Mn4+主导的EELS光谱证实(图3b)。这一结果表明,在高压范围内,岩盐相的形成是不利的。由于在如此高的电压下电解质的电化学氧化,只观察到薄CEI(<1 nm)。
当在WVR中循环时,表面演化得更加复杂。LNMO表面由1.5 nm厚的岩盐相和4 nm厚的低结晶度CEI外层组成(图3c)。因为WVR包括LVR,所以可以想象岩盐相在WVR中的形成就像在LVR中一样。岩盐相在WVR区(≈1.5 nm)比LVR区(≈7 nm)更薄,UVR区几乎不形成岩盐相。这些差异说明了低电压下岩盐相的自发形成和高电压下岩盐相的不稳定性。假设LVR中形成的岩盐相在充电到4.9 V等高电压时可以被分解。岩盐相分解为TM和O,引发电解液分解,形成CEI。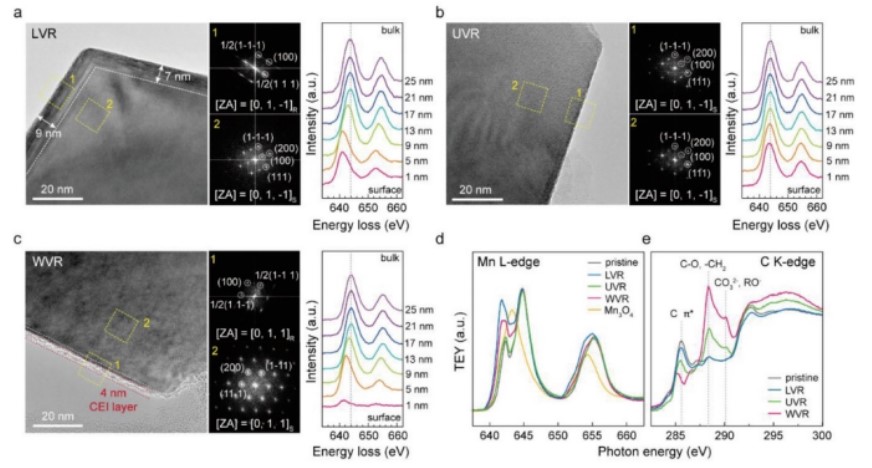
【图3】选定区域(10×10 nm2黄方区域)的HR-TEM图像和FFT模式与LNMO的空间分辨EELS光谱在a)LVR、b)UVR和c)WVR内进行100次循环后耦合。在LVR、UVR和WVR条件下,原始LNMO电极和LNMO电极在1 C下循环100次后的d)Mn L2、3-边和e)C K-边NEXAFS光谱。
近边缘X射线吸收精细结构(NEXAFS)光谱学提供了关于表面体系的化学状态的更详细的信息。
在总电子产率模式(TEY,探测深度=5-10 nm)下获得的Mnl边NEXAFS光谱可以比较原始LNMO、循环LNMO和Mn2.7+的Mn3O4中Mn的氧化状态(图3d)。
LVR中LNMO循环的NEXAFS光谱清楚地证实,由于岩盐相,Mn2+在表面占主导地位。与HR-TEM和EELS分析一致,岩盐相中Mn的还原量顺序为LVR>WVR>UVR=未循环LNMO。Ni L边NEXAFS光谱显示样品之间没有差异,因为尖晶石和岩盐相在3.5 V时都含有Ni2+。O K边缘TEY-NEXAFS光谱一致探测到边缘前区域的还原Mn2+阳离子,确认了LVR和WVR的表面岩盐相。
然而,O K边荧光产率(FY)NEXAFS光谱(探测深度为150-200 nm)显示,循环样品与原始LNMO之间的差异可以忽略,表明所有样品中都存在保存良好的大块尖晶石相。
C K边TEY-NEXAFS光谱提供了来自电解质分解的有机物种的信息。如图3e所示,在LVR中循环的LNMO的C K边NEXAFS谱与未循环的电极相差不大。这是由于电解液在2.3-4.3 V电压范围内具有较高的稳定性。
相比之下,在WVR中循环的LNMO光谱在288.3和290.1 eV处有强峰,分别对应于C-O、-CH2和CO32−、RO−物种。同时,由于CEI层覆盖了导电碳,在285.5 eV处的C π*峰强度降低。考虑到UVR和LVR中的CEI特征比WVR中的弱,得出结论,在WVR中循环会引发严格的电解质分解和CEI形成,而在高或低电压范围内循环则不会。
(3)高压下岩盐相的分解触发了CEI的形成
为了验证LNMO在WVR中表面岩盐相的演化,对LNMO的表面岩盐相进行了研究,LNMO在LVR中分别充电到3.5、4.3和4.9 V后,在99个循环中生长。图4a为第99个循环的电压分布图以及接下来的制样充电过程。Mn L边TEY-NEXAFS光谱(图4b)显示,含Mn2+的岩盐相在3.5和4.3 V时仍然存在。
然而,将LNMO充电到4.9 V会消除表面的Mn2+阳离子。结果,Mn4+阳离子主导LNMO表面。HR-TEM图像和FFT模式(图4c)进一步证实了高压驱动的岩盐分解,导致岩盐相厚度减少到2 nm。Mn的L边EELS深度曲线(间距为2 nm)也显示了Mn2+的表面存在。
同时,观察到在4.9 V充电过程后,C K边TEY-NEXAFS光谱中C-O(或-CH2)和CO3−(或RO−)峰增强(图4d),这表明岩盐相解离与CEI形成之间存在耦合。所有的实验证据都证实了MO型岩盐相在高压环境下的失稳。这一发现与之前的研究一致,即当充电电压超过4.6 V时,富镍层状正极中出现了表面岩盐和无序尖晶石相的高压驱动分解。
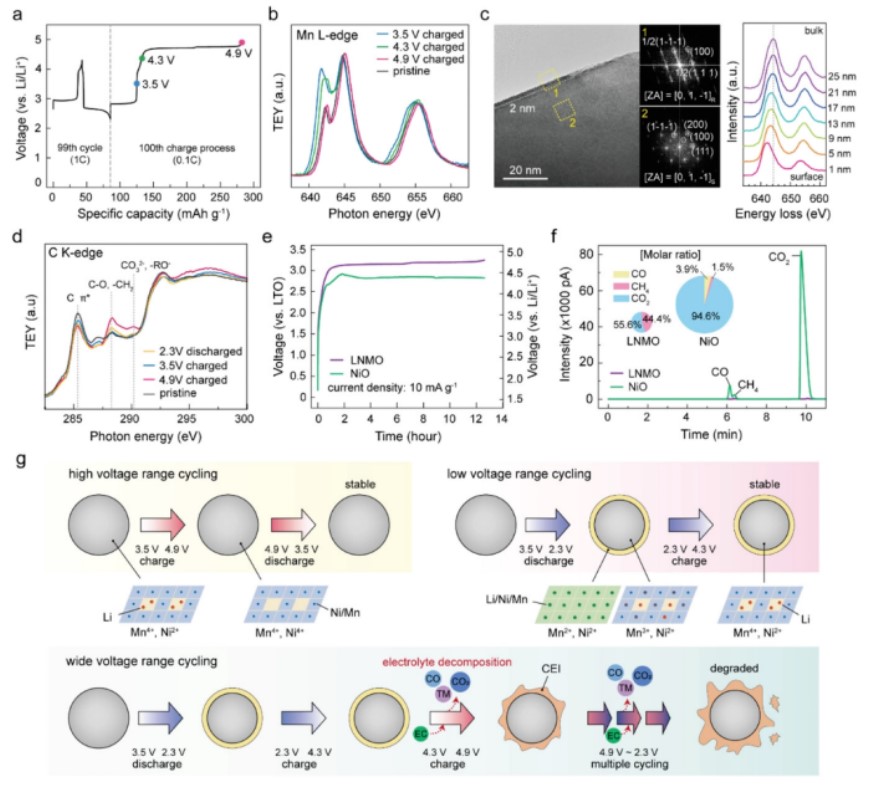
【图4】a)LNMO在第99和100个循环时的电压分布图。在99个循环中,LNMO电池在LVR中以1 C循环。在第100个循环时,正极在0.1 C下充电到4.9 V进行原位分析。b)充电至3.5、4.3和4.9 V时LNMO电极的Mn L2、3边NEXAFS光谱,如(a)所示。c)充电至4.9 V后,第100个循环LNMO选定区域(10×10 nm2面积黄色方框)的HR-TEM图像和FFT图。d)充电至3.5、4.3和4.9 V的LNMO电极的C K边缘NEXAFS光谱,如(a)所示。e)在电流密度为110 µA cm−2时,测量LNMO||LTO和NiO||LTO软包全电池的电压分布图,持续12h 36min。f)LNMO||LTO和NiO||LTO软包电池的气体演化GC-FID分布图。g)LNMO根据工作电压范围动态界面演化示意图。
为了建立岩盐分解与电解质降解之间的相关性,在恒电流条件下测试了岩盐型金属单氧化物NiO和MnO以及尖晶石型锰氧化物Mn3O4的电化学稳定性。
此外,还通过气相色谱-火焰离子化检测(GC-FID)对充电过程中收集到的软包电池内部气体进行定量分析,以全面了解复杂的分解反应。采用氧化钛锂(Li4Ti5O12,LTO)电极代替锂金属作为负极,以减少副反应作为比较,LNMO||LTO电池以相同的电流密度充电,持续时间相同。NiO电池在4.4 V vs Li(2.8 V vs LTO)时出现了一个长电压平台,表明岩盐相氧化(图4e)。
明显地,LNMO的氧化还原电位低于Ni2+/Ni4+的氧化还原电位。显示氧化是不可逆的,导致以下放电到2.3 V(vs Li)的容量可以忽略不计。可能存在于LVR内循环的LNMO表面的MnO和Mn3O4,在4.3 V(vs Li)以上也不容易氧化。这些结果证明LNMO正极在WVR中通过多阳离子氧化还原反应循环过程中反复形成岩盐并分解。
更重要的是,GC-FID定量结果表明,岩盐分解引发了显著的电解质分解(图4f)。尽管氧化电位较低,但NiO膜的气相副产物总量远远高于LNMO膜。主要成分为CO2和CO,分别占NiO电池释放气体的94.6%和3.9%。相比之下,LNMO电池在充电过程中释放的CO2气体少于NiO电池的1%,且没有释放CO气体,说明拓扑化释放反应有效地利用了大部分电子,没有发生明显的副反应。
NiO严格的气体演化行为导致软包电池膨胀,而LNMO电池保持其原始形状。在岩盐电解槽中受刺激的电解液分解的一个可行原因是过渡金属氧化物晶格在高电位下释放的活性氧(ROS)ROS,如单重态氧,会自发攻击碳酸乙烯等电解质溶剂,并启动开环反应,产生CH4、CO、CO2等各种气体,以及乙醇酸、草酸等可溶性副产物。副产物可以通过电化学和化学反应形成CEI层,进一步刺激电解质。证实,在WVR中循环时,LNMO正极释放的CO、CH4和CO2比在UVR中多192、2200和10倍。
综上所述,在WVR中,LNMO正极在低电压和高电压下经历了表面岩盐相的反复形成和分解,从而诱导了ROS的演化,导致电解质和电化学界面的持续恶化(图4g)。复杂副反应的积累导致电荷转移电阻的稳定增加和容量的快速下降。已知LNMO不太可能在5 V以下从晶格中释放ROS。因此,在常规条件下仅在UVR中循环的LNMO电池中,ROS和碳酸盐岩电解质之间不存在化学反应,仅确认了UVR中微量的气体生成。
在LVR中,尖晶石到岩盐表面的重建仅限于距离颗粒表面的7-9 nm范围内,并没有进一步进展到颗粒中心(图3a)。因此,可以预期LVR和UVR之间的转换频率可能会影响LNMO的寿命。为了研究电压范围变化频率对LNMO循环寿命的影响,每5个循环和50个循环循环LNMO交替LVR和UVR。
最终,LNMO在两种条件下都经历了100个LVR和100个UVR循环。在每5个循环的交流电压范围下,LNMO的容量损失更为严重。同时,LNMO在长期循环过程中出现了显著且持续的过电位增加。相比之下,在50次循环下,过电位几乎没有增加。
经过200个循环(100LVR+100UVR循环)后LNMO正极的EIS光谱显示,在更频繁的电压转变过程中,过电位的增加是由于正极-电解质界面的降解。降解阻碍了5循环蚀变过程中电荷转移反应的快速进行,Rct是50循环蚀变过程的3.3倍。
对相应的正极进行了X射线光电子能谱(XPS)分析,以追踪正极表面的CEI成分。在O 1s XPS光谱中,CEI组分(C-O、C=O、CO3和LiPFyOz之和)与TM-O的峰面积比分别为1.1、0.8和0.6,对于原始电极、50周电极和5周电极。与O 1s XPS光谱一致,C 1s光谱显示了5次循环改变电极中最主要的CEI组分。
得出结论,频繁交替电压范围会在CEI诱发电解质降解,阻碍电荷转移反应。WVR循环确实对应于LVR和UVR之间每一个循环的电压变化。因此,在低电压下重复岩盐形成,在高电压下分解,WVR发生了严重的降解,这反复导致ROS的演化和电解质的分解。
(4)通过修饰正极-电解质相互作用改善LiNi0.5Mn1.5O4的性能
提出的降解机制表明,调整正极-电解质相互作用的稳定性是实现LNMO正极在WVR中长期循环的关键。在调节界面稳定性时,发现对电解质配方进行简单的修改可以有效提高LNMO中多阳离子氧化还原的可逆性。图5a为LNMO在WVR中不同电解质的使用寿命;EC/DEC中的1 m LiPF6(1:1 v/v)(LP40),EC/DMC中的1 m LiPF6(1:1 v/v)(LP30),EC/DEC中的2 m LiPF6(1:1 v/v)(2 m LP40),DMC中的1 m LiPF6(1 m EC-free),DMC中的2 m LiPF6(2 m EC-free)。纯DEC溶剂与锂金属接触还原稳定性差,限制了DEC作为电解质中唯一溶剂的使用。
LP30的LNMO在100个循环中表现出50.9%的容量保持率,类似于LP40的情况下损失32.3%的容量。结果表明:在混合EC时,线性碳酸盐溶剂(DMC或DEC)的种类对LNMO的循环寿命影响不显著;在2 m LP40中,LNMO的容量比在1 m LP40中低16.9%,表明2 m LP40电解质的动力学缓慢。
从2 m LP40的离子电导率为1 m LP40的81.2%这一事实,将动力学迟缓归因于缓慢的界面电荷转移反应或脱溶反应。尽管容量较低,但2 m LP40的容量保持率提高了91.0%。这种改善可归因于溶剂化结构的改变,接触离子对增加,这可能影响CEI的组成和稳定性。
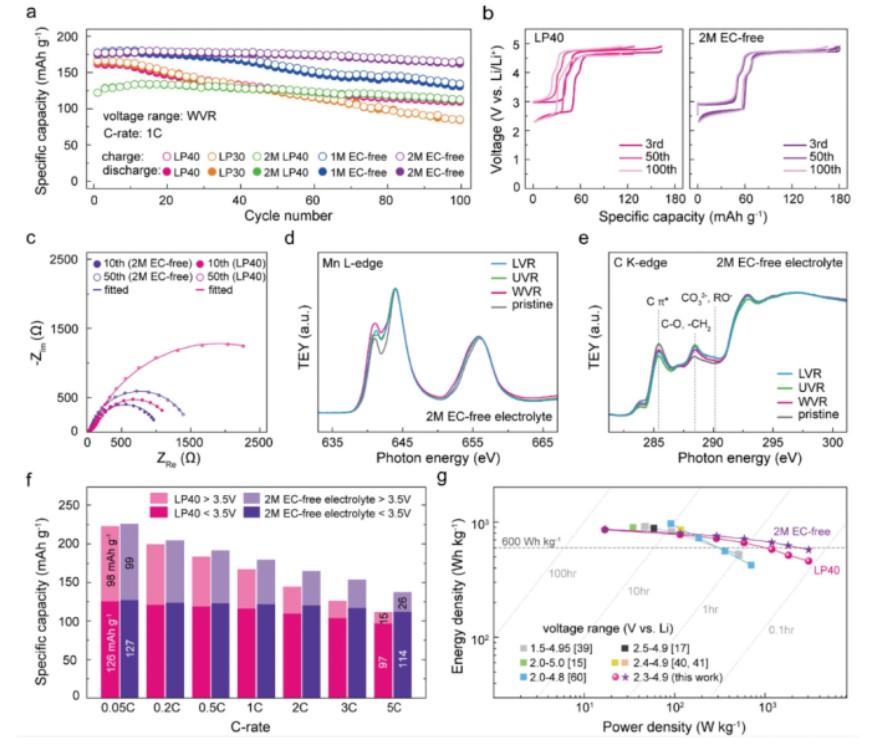
【图5】a)不同电解质组合LNMO电池的循环性能;1 C时WVR内LP40、LP30、2 m LP40、1 m 无EC和2m无EC电解质。b)1 C时WVR内LP40和2 m无EC电解质的LNMO的第3、50和100电压分布图。c)在WVR中添加LP40和无EC电解质的LNMO半电池的第10和第50EIS谱。EIS在2.3 V下测量。d)Mn L2,3边和e)C K边NEXAFS光谱,原始LNMO电极和使用无EC电解质在LVR,UVR和WVR条件下在1 C下循环100个循环的LNMO电极。f)根据C倍率,LNMO与LP40和无EC电解质的实际容量。g)显示利用多阳离子氧化还原的LNMO的Ragone图。对角线对应LNMO单次充电或放电过程所需时间。
在DMC中1 m的LiPF6中,LNMO稳定地保持了最初30个循环的容量,然后开始失去它,表现出分解过程。因此,将传统电解质中的ROS演化归因于EC从TM氧化物中提取氧气的催化作用除ROS外,界面的动态演化和岩盐相分解也会加速TM在电解液中的溶解。对来自循环电池的电解质的ICP-OES分析证实,TM溶解在LP40中比在无EC电解质中更为严重。
溶解的Mn2+更倾向于与EC溶剂化,导致负极和固态电解质界面在100个循环过程中的降解,过电位在无EC电解液中保持较小,而在LP40和LP30中迅速增加(图5b)。当活性材料负载较高(≈5.6 mg cm−2)时,含LP40的LNMO的过电位增加和容量损失加快。
通过长期循环伏安法观察LNMO在商用电解质中的显式降解。LP40中过电位的增加导致了Mn3+/Mn4+氧化还原能力的降低。从LNMO正极经过100 CV循环放电至2.3 V的XRD谱图可以看出,无EC电解质中的四方相比LP40更明显。考虑到在无EC电解液中有明显更好的容量保留(图5a),该结果否定了相变程度与容量衰减之间的直接相关性。相反,界面处的退化可以延缓体相转变。
将LNMO在无EC电解液中循环寿命的延长归因于Rct的抑制增长,如EIS光谱(图5c)所示。在LP40中,Rct在第10个循环后的40个循环中增加了263%。相比之下,在无EC电解液中仅观察到76%的Rct增加。结果表明,与LP40相比,无EC电解液具有更稳定的电化学界面,使循环时间更长,容量更大。
Mn L边TEY-NEXAFS谱阐明了电解质修饰的结果(图5d)。在所有电压范围内,在无电解液中循环后,在LNMO表面几乎不存在3.5 V(vs Li)的二价锰阳离子,这表明表面重建到岩盐相的减少。此外,C K边TEY-NEXAFS光谱显示,C-O、-CH2和CO32−、RO−峰分别在288.3和290.1 eV处小幅增加,表明电解质分解有所缓解(图5e)。结果表明,EC是触发正极电化学界面严重退化的主要电解质成分,需要在比传统电压范围更宽的电压范围内循环。此外,结果还证实了岩盐形成/分解、电解质分解和正极中CEI形成之间的强耦合。
界面修饰不仅延长了LNMO的循环寿命,还为其利用多结氧化还原提供了更高的速率能力。在无EC电解液中,LNMO正极在0.05、0.2、0.5、1、2、3和5 C下分别提供226、205、192、180、166、154和139 mAh g−1的容量。在LP40中,容量始终较小,并且在较高的倍率下差异变得更大。
电压曲线表明,在无EC电解液中,Ni和Mn氧化还原平台的过电位都低于LP40。在电荷转移动力学增强的情况下,商业LNMO在0.05、0.2、0.5、1、2、3和5 C下的能量密度分别为866、801、758、715、665、624和574 Wh kg−1。这些值分别比LP40大1.1%、3.1%、5.2%、8.1%、14.7%、21.3%和25.3%。无EC电解质对应的功率密度分别为17、116、292、588、1188、1796和3034 W kg−1,如图5g中的Ragone图所示。在WVR的高C倍率(3C)下,LNMO正极在2 m无EC电解液中经过500和700循环后的容量保持率分别为71.5%和57.5%,而在LP40中经过500循环后的容量保持率为20.1%。
注意到,由于在DMC中2 m LiPF6中的石墨负极具有较高的电化学稳定性和倍率能力,LNMO正极可以与预锂化石墨负极耦合,使用无EC电解质形成全电池。LNMO|2 m LiPF6在DMC|石墨全电池中在3 C时表现为119 mAh g−1,200次循环后保持71.5%的容量。据所知,在使用多阳离子氧化还原反应时,LNMO具有如此优异的功率密度和寿命是前所未有的。
尽管在一些研究中,通过调整二次颗粒形态和额外的热处理,已经实现了循环寿命的延长,但由于活性界面的减少,增大的颗粒尺寸在很大程度上限制了倍率性能。
与Ni2+/Ni4+电压平台相比,随着倍率的增加,Mn3+/Mn4+电压平台的缩短更为显著(图5f)。注意到动力学随电解质配方、颗粒尺寸和表面积的不同而有很大的变化,这无疑证实了促进界面电荷转移对于最大限度地提高LNMO的能量和功率密度并实现长循环循环至关重要。除了本研究中展示的电解质修饰外,相信通过包覆层、掺杂和面控的表面工程将进一步提高用于下一代RLB的LNMO性能。
四、总结与展望
综上所述,揭示了LNMO在大电势变化(>2.5 V)驱动下的动态表面重建,重建了同时使用Ni和Mn氧化还原进行高能存储时LNMO的失效机制。低压时LNMO表面转变为岩盐结构,高压时岩盐层解离,电解液分解。重复的界面演化导致电荷转移阻抗的快速波动,导致循环过程中的容量衰减。相反,发现在正极电位低于4.3 V的条件下,Mn3+/Mn4+氧化还原反应具有良好的可逆性,这表明有必要重新审视高估了Jahn-Teller畸变后果的传统降解理论。最后,证明了电解质配方是提高LNMO界面稳定性、抑制尖晶石向岩盐转化的有效策略。稳定的电化学界面使LNMO在可循环性、可逆容量和功率密度方面具有优于商业电解质的优良性能。发现了界面对LNMO循环的显著作用,这为开发高能和长循环寿命的富锰化学RLB正极材料(包括尖晶石、富Li/ Mn层状和无序岩盐氧化物)提出了一种有前景的策略。阐明和调节锰氧化物表面潜在依赖的结构稳定性也将指导合成和催化剂的研究。
文章二:梯度释氧缓冲和界面离子自加速稳定高压富镍正极的原位构建
一、全文概要
具有优越能量密度的富镍正极引起了锂离子电池(LIB)的广泛关注,但其商业化受到结构退化、热失控和急剧容量衰减的阻碍。来自河南大学的白莹团队采用了一种简单的原位构建策略,将具有高结合能的硼(B)梯度地嵌入到一次颗粒中,并将压电Li2B4O7(LBO)均匀沉积在多晶LiNi0.8Co0.1Mn0.1O2(NCM811)表面的二次颗粒上,旨在同步提高循环后的电化学稳定性和Li+动力学。
特别是,LBO改性的NCM811正极在锂金属负极下,具有极好的容量保持率(300次循环,1 C后为88.9%)和倍率性能(112.2 mAh g−1,10 C),NCM811 LBO/Li4Ti5O12全电池在1000次循环(0.5 C)后的容量保持率为92.6%。多尺度原位/非原位表征和有限元分析表明,LBO改性的改善机理是合理设计的O释放缓冲剂和界面阳离子自加速的协同作用。本研究为防止高能LIB的结构退化和热失控提供了一种简便实用的方法。
研究以题目为“In Situ Construction of Gradient Oxygen Release Buffer and Interface Cation Self-Accelerator Stabilizing High-Voltage Ni-Rich Cathode”的论文发表在材料领域顶级期刊《Advanced Functional Materials》。
二、研究亮点
1、通过原位构建方法,将与O具有高结合能的硼(B-O:809 kJ moL−1)梯度加入到每个一次颗粒中,并将压电Li2B4O7同时修饰在LiNi0.8Co0.1Mn0.1O2表面。
2、通过大量的原位/非原位实验研究和理论计算,全面阐明了B阳离子在富镍材料中的掺杂位置及其对稳定晶格结构的作用。
3、作为一种典型的压电材料,沉积的LBO化合物在循环过程中通过引入局域极化电场,有效加速了界面Li+扩散动力学。理论计算和有限元分析进一步阐明了通过B3+梯度掺入和LBO表面改性,NCM811表面离子扩散能垒降低,应力积累减弱。
4、LBO改性的NCM811正极具有优异的容量保持性能。该工作不仅为构造具有良好结构相容性和稳定性的均匀包覆层提供了一种简便实用的策略,而且为抑制超高电压富镍正极中的氧释放开辟了新的途径。
三、图文导读
为了阐明B掺杂对富Ni样品中晶格O的影响,进行了详细的理论分析,结果见图1。
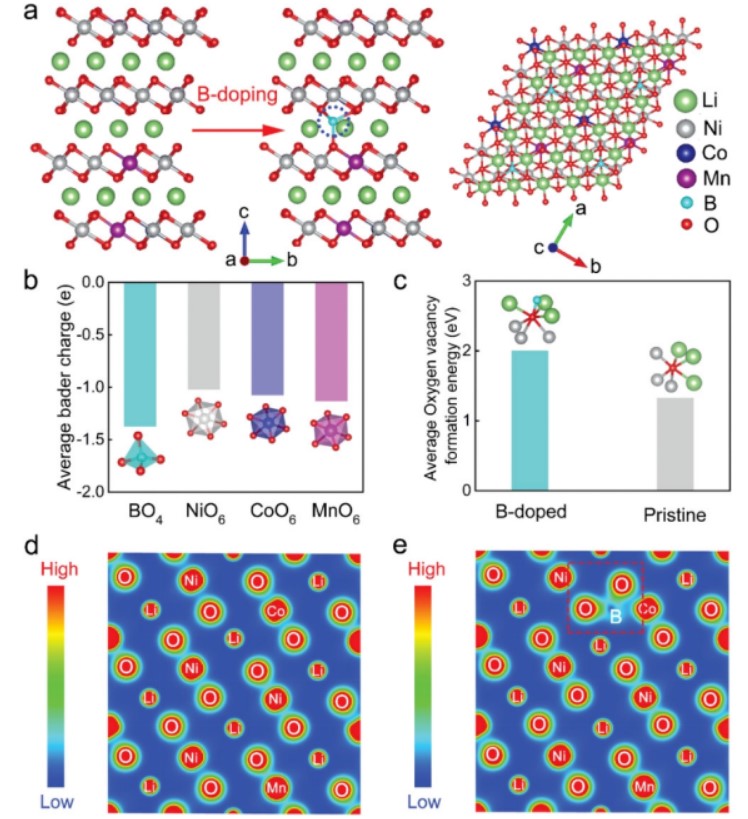
【图1】a)NCM811和NCM811-LBO的晶体结构示意图;b)BO4四面体和MO6八面体(M=Ni,Co,Mn)中O原子的Bader平均电荷;C)平均氧空位形成能;d)NCM811和e)NCM811-LBO的电荷密度等值线图。
进一步对原始NCM811和NCM811-LBO样品的O空位形成能量进行量化,揭示了B掺杂对O晶格的稳定作用。
用LBO通过简单的一步退火方法对改性NCM811正极进行原位改性的策略如图2a所示。此外,利用压电力显微镜(PFM)研究制备的LBO材料的压电性能(图2h)、高对比度振幅和相位图像(图2h)显示了理想的压电效应采用聚焦离子束结合扫描电子显微镜(FIB-SEM)技术对合成的NCM811-LBO二次颗粒的截面元素分布进行了研究。图2b和对应的截面结构能谱(EDS)映射明显表明,在800 C的高温下,H3BO3(HBO)可以原位岩性化成具有快速Li+导电性和压电性能的LBO。同时,在热驱动过程中,B3+阳离子会梯度地结合到每个一次颗粒中。
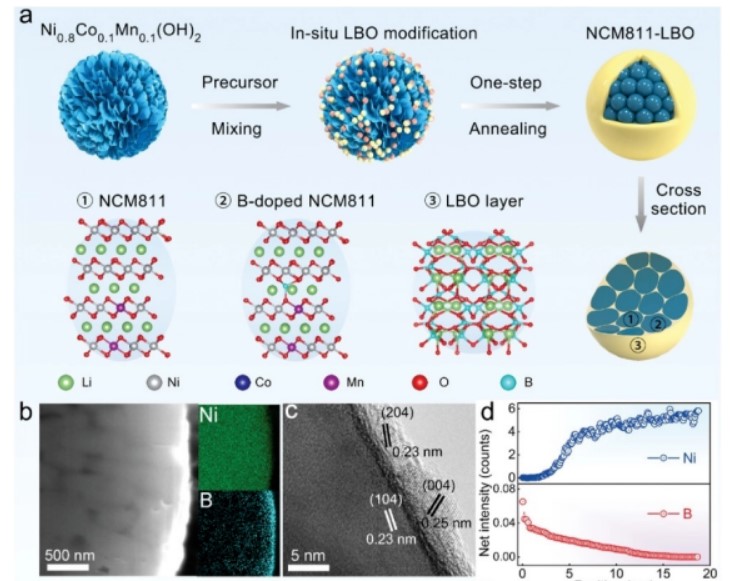
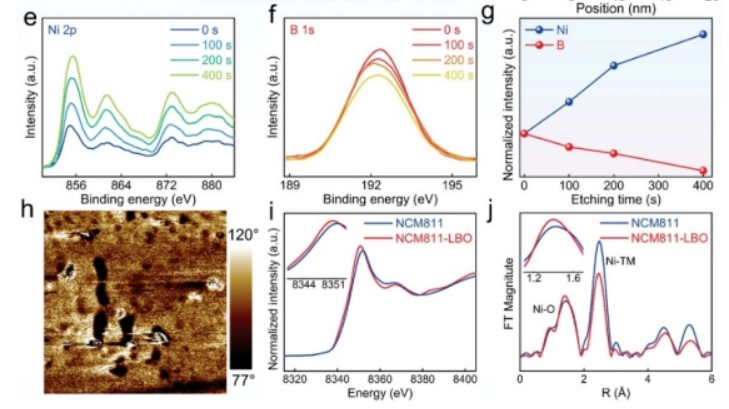
【图2】a)NCM811-LBO的制备原理图;b)NCM811LBO带元素映射(Ni和b)的截面SEM图像;c)HRTEM图像和d)NCM811-LBOTEM-EDS线扫描结果;不同氩离子刻蚀时间下e)Ni 2p,f)B1s,g)对应归一化结果的XPS谱;h)PFM技术检测LBO的相位图像;i)NCM811和NCM811-LBO的XANES和j)Ni边EXAFS谱。
采用高分辨率透射电子显微镜(HRTEM)对NCM811LBO的微观结构进行了表征。如图2c所示,NCM811-LBO表面沉积了一层厚度≈5 nm的非均匀致密层。随着线扫描深度的增加,Ni元素的强度有所提高,B信号有所减弱(图2d,TEM-EDS线扫描图像),从另一个角度说明了NCM811-LBO中LBO封装和B梯度融合的成功。观察到LBO层的厚度为≈5 nm(图2c),TEM-EDS线扫描图像显示,B信号从二次颗粒表面延伸到晶格内部≈15 nm的深度,因此B离子在晶格表面到晶格内部≈10 nm的范围内进行梯度掺杂。
采用X射线光电子能谱(XPS)表征来检测制备样品的元素化学环境。采用XPS深度分析检测了NCM811-LBO颗粒的Ni和B分布,并对其进行了连续的氩离子刻蚀。如图2e-g所示,随着刻蚀时间的延长,B信号从表面向内体逐渐减弱,同时Ni元素的峰值持续增长,为B阳离子的梯度掺杂提供了有力的证据。
为了揭示b掺杂对Ni元素价态的影响,采集了X射线吸收近边结构(XANES)光谱,归一化结果如图2i所示。通过扩展X射线吸收精细结构(EXAFS)光谱的傅里叶变换幅值得到Ni边缘的局部配位信息(图2j)。
首先,对不同LBO处理策略下的NCM811电极进行半电池测试,并进行比较。原位LBO修饰的NCM811比其他电极表现出更好的容量保持能力,证实了在电化学性能方面的协同优势。图3a显示,NCM811-LBO正极在0.1 C(22 mA g−1)下的初始放电容量为2012 mAh g−1,比其原始正极(194.6 mAh g−1)高约6.6 mAh g−1。
比容量的增加可以归因于镍价的降低,为B掺杂到四面体位置提供了另一个有力的证据。从图4b-d可以看出,改性富镍正极的电化学性能明显提高。在25 C下循环300次(图3b),NCM811-LBO正极仍能提供168.2 mAh g−1的可逆容量,在1 C下容量保持率为88.9%,而在相同的电化学过程中,原始的NCM811仅保持77.6 mAh g−1的容量和42.1%的容量保持率。
NCM811-LBO正极在电流密度为10 C时,放电容量高达112.2 mAh g−1(图3c),远高于NCM811材料(78.6 mAh g−1)。此外,NCM811-LBO/Li4Ti5O12全电池在0.5 C下循环1000次后仍保持了92.6%的优异容量(图3d)。电化学性能和倍率能力的增强同时说明原位LBO处理改善了结构稳定性和Li+输运动力学。
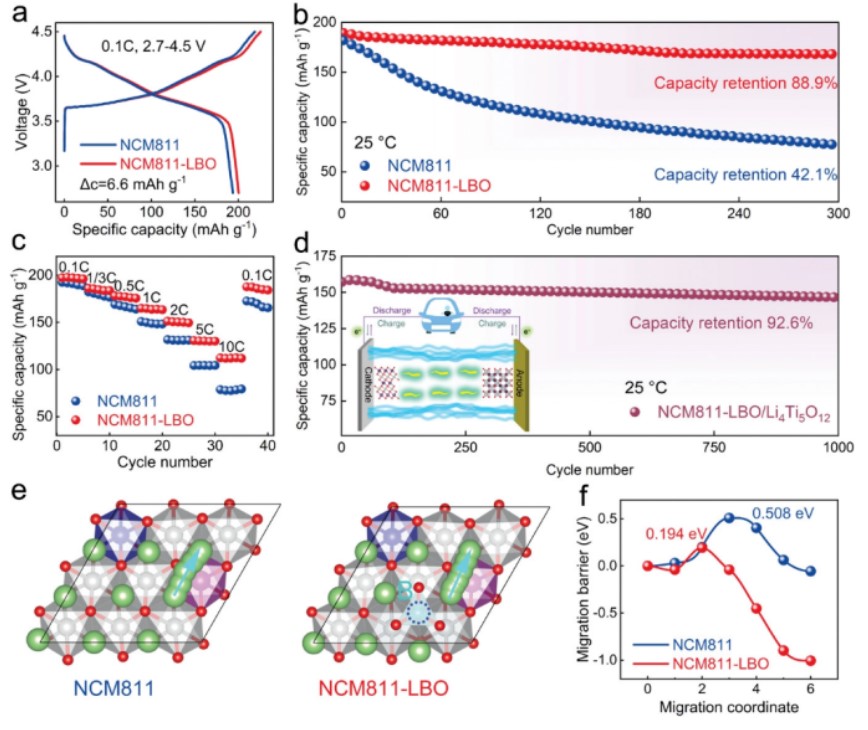
【图3】a)NCM811和NCM811-LBO的初始充放电分布;b)NCM811和NCM811-LBO在25°C下1 C的循环性能;c)NCM811和NCM811-LBO的倍率性能;d)NCM811-LBO/Li4Ti5O12全电池在0.5C、25 C条件下的循环性能;e)Li+扩散路径的理论模型和f)NCM811和NCM811-LBO对应的扩散能垒。
保持良好的结构稳定性和增强的相变可逆性可以部分解释为dQ/dV曲线。采用密度泛函理论(DFT)计算方法分别研究了NCM811和NCM811-LBO正极的Li+扩散势垒。基于Li+在大块富Ni材料中的迁移路径理论模型(图3e),表明B掺入后可获得314m eV的扩散势障下降(图3f),这在一定程度上解释了CV和EIS分析中Li+扩散动力学的增强以及倍率性能的改善(图3c)。
此外,为了直接揭示Li2B4O7对富镍正极Li+动力学的任何压电效应,研究了不同扫描倍率下的CV曲线以及原始NCM811、非压电LBO-NCM811和压电LBO-NCM811的倍率性能,LBO处理后电极极化得到了有效的缓解,特别是对压电LBO修饰电极。拟合的CV曲线为压电LBO处理增强Li+动力学提供了明显的证据。此外,压电LBO修饰的NCM811正极的倍率性能也表现出最有效的改善,再次为Li2B4O7的压电效应在循环后促进富镍正极的Li+动力学提供了坚实的证据。
采用原位差分电化学质谱(DEMS)检测初始循环后NCM811和NCM811-LBO电极的不可逆氧氧化还原活性(图4a,b)。在原始NCM811的锂化过程中,可以显著研究到O2和CO2的释放(图4a),而NCM811-LBO正极的相应曲线中没有明显的气体释放(图4b)。
在4.0V~4.5 V的高电位下,检测到的CO2可能与碳酸盐岩杂质(如Li2CO3)的分解有关,而收集到的O2则与晶格氧的不可逆氧化和逸出有关重复循环过程中有害的氧气释放会导致不可逆的结构转变和快速的性能衰退,以及严重的安全问题。本文的结果清楚地证实了LBO修饰可以稳定富镍正极中的晶格O,与上述理论计算一致(图1b-e)。
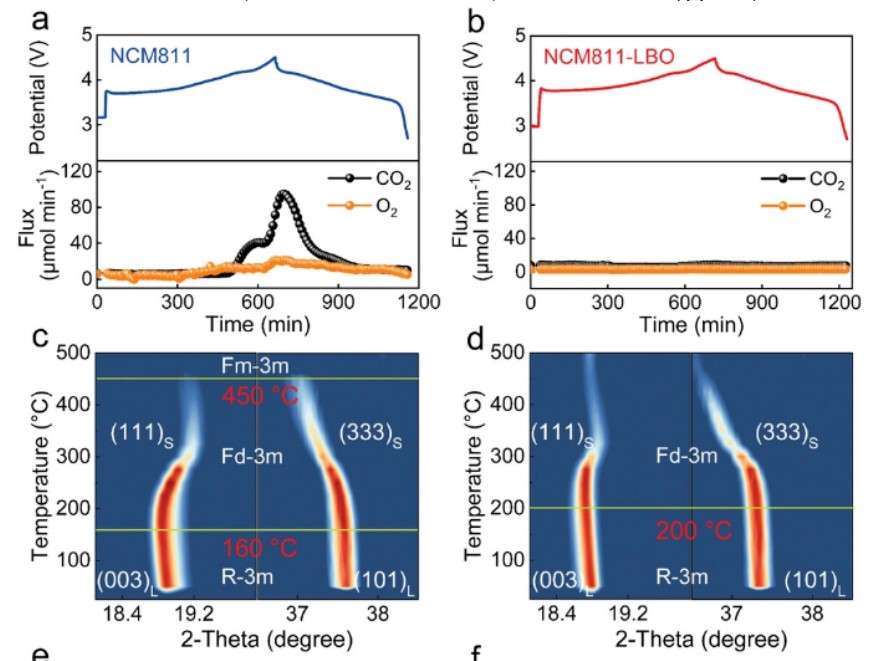
【图4】a)NCM811和b)NCM811-LBO的原位DEMS测量;c)NCM811和d)NCM811-LBO的原位HT-XRD图谱;e)c和f)a晶格参数的归一化演化。
通过原位高温XRD(HT-XRD)表征,确定了脱锂态电极的结构和热稳定性(图4c-f)。相变延迟证实了NCM811-LBO电极增强了结构/热稳定性,降低了氧活性。为了定量揭示晶格在加热过程中的变化,进一步通过Rietveld细化XRD谱图得到晶格参数(图4e,f)。
初始点阵参数c的轻微增长可归因于适度处理下的点阵膨胀,而后者则与相变和氧释放有关相比之下,NCM811-LBO正极的晶格参数c在退化前表现出更大的膨胀,证实了NCM811-LBO正极比原始材料能够抑制更多的c轴膨胀。图4f中晶格参数a在200 C以下的线性膨胀主要是由于热膨胀。虽然随着温度的进一步升高,由于TM离子的迁移和相应的相变,a轴急剧升高,但改性电极的相变温度明显升高。
原位HT-XRD研究到的NCM811-LBO样品的相变和晶格变化明显减弱,表明LBO处理有效地缓解了氧释放和热失控,与原位DEMS测量结果一致(图4a,b)。
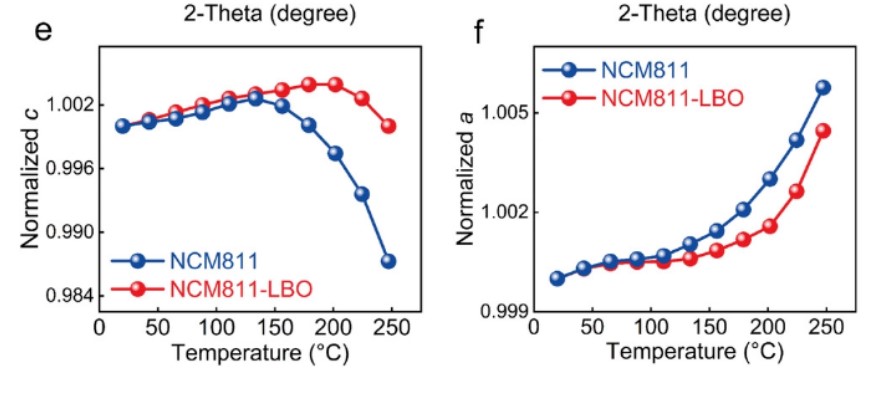
在初始循环中进行原位XRD分析,以研究原位LBO对结构稳定性的影响(图5a,b)。晶格参数c经历了逐渐增大和急剧退化的非线性演化过程(图5c)。
【图5】a)NCM811和b)NCM811-LBO初始循环(003)峰的原位XRD图;c)NCM811和NCM811-LBO晶格参数c的演化;d)用PFM法测定NCM811的杨氏模量;e)NCM811-LBO初始循环应力曲线;f)LBO层压电场分布的有限元分析;g)压电LBO层对NCM811界面Li+输运的调节作用示意图。
用原子力显微镜(AFM)检测原始NCM811的杨氏模量为109 GPa(图5d)。NCM811-LBO在初始循环中的应变和应力值由公式计算得到,如图5e所示。随着内部富Ni颗粒晶格应变的传递,压电LBO层上的感受力会激发附加的极化电场,如图5f所示。在NCM811-LBO正极中触发的极化电位最大值在带电状态下为0.52 V。在放电过程中,可以获得相同的最大电位,在第1次循环结束时保持0.24 V的额外电压(图5f)。
基于以上分析,LBO对富镍电极的贡献示意图如图5g所示。原始NCM811中的有害应变会导致内部微裂纹延伸到外表面,其中渗入的电解液不可避免地会对电极材料造成严重的侵蚀。但LBO压电层通过合理地改变NCM811的应变-应力发挥了积极的作用,特别是在循环过程中,LBO化合物中诱导的极化电位与外部电场的极化电位方向一致,在电解-正极界面上起到局部加速器的作用,促进Li+的扩散。同时,改进的Li+动力学可以通过降低界面能垒来降低应力-应变积累,最终提高富镍正极的电化学性能。
图6a-d采集了NCM811和NCM811-LBO正极第一循环的原位拉曼光谱。对NCM811和NCM811-LBO二次颗粒在不同电压下的应力-应变分布进行了有限元分析,截面映射如图6e、f所示。可见,NCM811正极具有较大的应力分布,而NCM811-LBO二次颗粒明显缓解了这一应力分布这种差异主要是由于LBO的原位构造,通过独特的压电效应有效地缓解了富镍正极的应力积累。
四、总结与展望
本研究将对O晶格具有高结合能的非均相B3+离子梯度加入到每个一次颗粒中,并将压电LBO层封装在多晶NCM811正极的二次颗粒表面,旨在通过一种简便的原位构建方法同步改善其结构和电化学性能。制备的NCM811-LBO电极在1 C的电流密度下,经过300次循环后仍能保持88.9%的初始放电容量,明显优于原始电极的63.2%。此外,在0.5 C条件下,NC811-LBO/Li4Ti5O12全电池在1000次循环后仍能保持92.6%的容量。结合理论计算和有限元分析的多尺度原位实验表征表明,LBO改性样品具有良好的结构稳定性。改进后的正极循环性能和结构稳定性显著提高,主要归功于以下协同作用:1)加入B3+阳离子有效降低了TM-O共价和O原子电负性,抑制了晶格O逃逸;2)B离子独特的掺杂位置(锂层中的四面体位置)在高析出的富镍正极中具有柱效应,有效稳定了晶体结构;3)原位压电LBO修饰物在循环过程中减轻了内部材料的有害应力,加速了锂离子在正极-电解质界面的扩散;4)沉积的LBO层作为物理分隔层,通过减轻电极和电解质之间可能发生的副反应来改善界面的不稳定性。本文对NCM811正极进行梯度掺入和均匀压电LBO修饰的同步改性,不仅对操控特高压富镍正极的内部结构和界面稳定性具有指导意义,而且为能量储存和转换系统的设计和开发开辟了新的思路。
审核编辑:刘清
-
锂电池供电威廉希尔官方网站 保护方案2024-04-16 0
-
单片机如何设计锂电池充电器2011-11-30 0
-
纳米氧化镁应用在电池中有什么效果2015-04-08 0
-
磷酸铁锂电池的特性解析与电源管理芯片的应用2015-10-13 0
-
磷酸铁锂电池的八大优势2015-10-17 0
-
磷酸铁锂电池的优缺点及行业应用2017-02-15 0
-
蓝博科普 你知道电池都有哪些种类吗?2018-12-22 0
-
可充电的锂电池为什么能得到广泛使用?2021-03-11 0
-
锰酸锂电池优缺点_锰酸锂电池充电次数2020-04-14 42705
-
锰酸锂电池的技术参数_锰酸锂电池能量密度2020-04-14 14460
-
宁德时代或将推出不含镍钴锰的锂电池?2020-09-06 3034
-
锂过量的正极材料中可逆的Mn2+/Mn4+双氧化还原2020-12-25 785
-
什么叫做三元锂电池?详细介绍三元锂电池的优点和缺点2022-05-20 11806
-
卡拉胶作为5V高压LiNi0.5Mn1.5O4正极牺牲粘合剂!2023-08-01 1210
全部0条评论

快来发表一下你的评论吧 !
