

4.2V高压全固态聚合物电解质新突破
描述
研究背景
聚氧化乙烯(PEO)固体电解质(SE)在全固态锂电池(ASSLB)中是可行的,并具有驾驭电动汽车的高安全性。然而,普通PEO固体电解质具有低氧化分解电位(低于4V),在高压下极易分解,以致于基于PEO的ASSLBs的高电压(≥4.2V)正极材料无法使用,牺牲了电池的能量密度。
成果简介
近日,湖南大学刘继磊教授在浓盐化学的基础上,调节PEO电解质中锂盐的浓度,发现高浓度PEO SEs(EO:Li+ ≤ 6:1)拥有高氧化电位(>5 V vs. Li/Li+),氧化电位随配位环氧乙烷程度的增加而不断上升。因此,其团队设计了在正极侧有PEO(EO:Li=4:1)和负极侧PEO(EO. Li=16:1)的双层SE,可以与4.2V级别的LiCoO2和LiNi0.6Co0.2Mn0.2O2正极材料相匹配,使用该双层SEs的ASSLBs在充电到4.2V和4.4V时表现出更稳定的循环性能。Li//LiCoO2 ASSLB的初始充电/放电容量为143.5/131. 5 mAh g-1,速率为0.1 C,截止电压为4.2 V,100次循环后放电容量保持在82.6%左右,优于使用EO:Li=16:1(PEO(16:1))的SE的电池。
研究亮点
1、发现氧化电位强烈依赖于配位的EO链,并随着盐浓度的增加而增加,最高值为>5V(当EO:Li+≤6:1)。 2、提出了在正极侧使用PEO(EO:Li=4:1)和负极侧使用PEO(EO:Li=16:1)的双层SE,来实现4.2V高压全固态PEO基电池。 3、通过高浓度设计,形成薄而均匀的CEI层,有效抑制了界面降解动力学、CEI中的电子传导以及LiCoO2到Co3O4的不可逆相变。
图文导读
由图1a可见,在3.9V附近,PEO SE氧化产生的电流增长可以忽略不计,而高浓度PEO的电流增长则更弱,作者将氧化阈值电位定义为电流密度达到7.7 μA cm-2但不上升时的电位,对于PEO(16:1)、PEO(10:1)和PEO(4:1)SEs,它们分别为4.68 V、4.81 V和5.19 V。涉及Li+、TFSI-和EO链的配位结构是决定氧化电位的关键因素,图1b中位于1063、1096、1108、1149和1119 cm-1的峰是自由环氧乙烷链,而位于1056、1086和1135 cm-1的峰则是协同环氧乙烷链。1056 cm-1处的峰有明显的蓝移,1119 cm-1处的峰有明显的红移,这分别是由于环氧乙烷与离子之间的相互作用增强和无定形相含量增加造成的。
因此,1056、1086和1135 cm-1处的峰的聚集面积与所有与环氧乙烷有关的峰的相对值被用来描述环氧乙烷链的协调程度,它随着EO:Li变成6:1而逐渐增加,然后保持不变,直到EO:Li达到4:1(图1c),表明几乎所有EO链参与配位结构,因此锂盐超过6:1(临界浓度,图1c)的PEO SE被定义为高浓度的SE。仔细研究配位的EO程度对氧化电位的影响,可以发现氧化电位持续增加(图1d),而在临界浓度之后,电导率明显下降。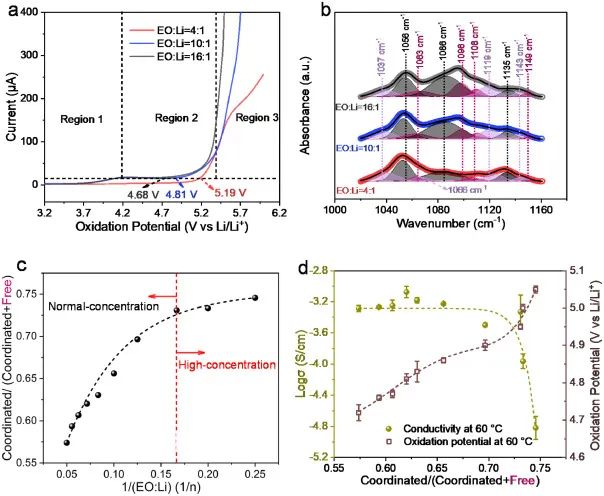
图1. PEO SEs的电性能和配位结构。(a) PEO SEs的LSV曲线,速率为0.5 mV s-1。(b) 在室温下获得的PEO SEs的FTIR光谱和去卷积结果。(c) 根据FTIR光谱计算的PEO链中配位环氧乙烷与总环氧乙烷的含量。(d) 配位环氧乙烷的程度对离子电导率和氧化电位的影响。
图2a描述了Li/PEO(16:1)/PEO(4:1)/LiCoO2 ASSLBs的组装情况,以评估高浓度PEO SEs与高压正极耦合的实用性,Li/PEO(16:1)/LiCoO2 ASSLBs也被作为对照样品进行研究。在60℃和0.1C下,Li/PEO(16:1)/PEO(4:1)/LiCoO2 ASSLB在4.2V截止电压下的初始充电/放电容量为143.5/131.5 mAh g-1(图2b),而在4.2V截止电压下的放电容量约为理论值的93.2%。
在100个循环之后,放电容量仍然是108.6 mAh g-1,约为初始容量的82.6%(图2c)。图2d表明双层SE也能使Li/NMC ASSLBs提供令人满意的性能。值得注意的是,与文献中ASSLB的电化学性能相比,该工作结果具有竞争力(图2e)。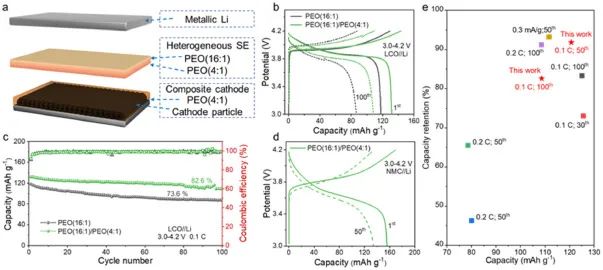
图2. 4.2V级ASSLBs的电化学性能。(a) 使用PEO(16:1)/PEO(4:1)SE的ASSLBs示意图。(b, c) 使用PEO(16:1)和PEO(16:1)/PEO(4:1)SE的Li/LCO (LiCoO2) ASSLBs的循环性能。(d) 使用PEO(16:1)/PEO(4:1)SE的Li/NMC ASSLB的循环性能。(e) 本工作与文献中ASSLB的电化学性能比较。
SE和LiCoO2界面降解形成CEI层是性能下降的主要原因。作者用Wagner模型研究界面反应动力学,它基于监测在60℃下以0.1C的速率充电到预定电压后静止时间的阻抗演变。对于充电到4.2V的ASSLB(图3a),在阻抗谱中有两个半圆,因此使用等效威廉希尔官方网站
对它们进行拟合,如图3a的插图。Rs可以归因于SE的电阻。
在不同的静止时间后,仔细观察第一个半圆,其数值几乎相似(图3a),因此第一个半圆可以归因于Li/PEO界面(RSEI),因为PEO SE对金属Li拥有良好的电化学稳定性。但是第二个半圆随着静止时间的增加而增加(图3a),它可以归结为PEO/LiCoO2界面的RCEI。值得注意的是,PEO(4:1)/LiCoO2界面的RCEI比PEO(16:1)/LiCoO2界面的RCEI低(图3b,c),这有利于界面电荷运输,尽管前述SE的电导率较低。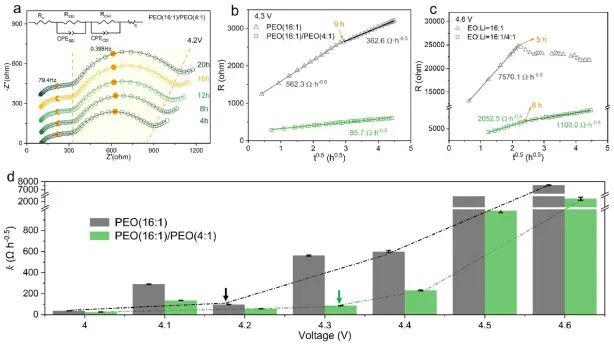
图3. 界面降解动力学分析。(a) 使用双层SE的Li/SE/LiCoO2 ASSLB在充电到4.2V后静止状态下的阻抗光谱。(b, c) RCEI随t0.5的变化和(d) 使用PEO(16:1)/PEO(4:1)SE的Li/SE/LiCoO2 ASSLB的瓦格纳型模型计算的k。
图4a中有一个位于675 cm-1的振动峰,可以归属于Co3O4的A1g振动模式。另外两个位于485 cm-1和595 cm-1的峰分别归属于LiCoO2的E1g振动模式和A1g振动模式 。此外,Co3O4和LiCoO2的A1g模型的整合区域被用来分析Co3O4的分布。在与PEO(16:1)/PEO(4:1)SE耦合的正极中,Co3O4的数量较少且分布在局部(图4c),而在与PEO(16:1)SE耦合的正极中,它的数量丰富且分布广泛(图4d)。
Co3O4是由LiCoO2在循环过程中的不可逆相变引起的。一方面,强化的不可逆相变直接加速了使用PEO(16:1)SE的电池的容量衰减。另一方面,处于高氧化状态的Co3O4可以加速PEO SE的氧化,这进一步引起了容量衰减,而PEO SE的高浓度设计可以抑制LiCoO2的不可逆相变。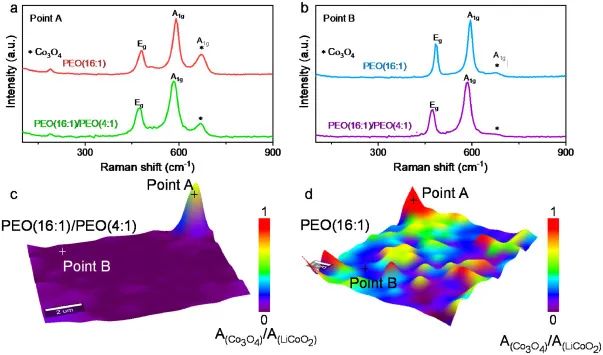
图4. 钴酸锂正极的相位变化。使用(c)PEO(16:1)/PEO(4:1)和(d)PEO(16:1)SEs的ASSLBs的正极的拉曼(a, b)光谱和(c, d)映射在20个循环后的检索。
图5a-f显示了循环后正极中F、S和O元素的分布,在使用PEO(16:1)/PEO(4:1)SE的ASSLB中,富含F的区域是极少数,富含S的区域几乎没有发现(图5a和b)。富含F和S的区域较少,这可能是由于浓盐设计抑制了锂盐的界面降解。作为参考,在使用PEO(16:1)SE的ASSLB的LiCoO2上,富含F(图5d)和富含S(图5e)的区域是明显的,这种富集是由锂盐的显著降解造成的。
应该指出的是,富含S的区域可能是多硫化物,导致CEI层的电子传导显著。此外,在与PEO(16:1)/PEO(4:1)SE结合的LiCoO2表面上,与PEO(16:1)SE的情况相比,由PEO基体的氧化引起的富O区域也是边缘形容词(图5c和f)。因此,参照原始的LiCoO2(图5g),在含有PEO(16:1)/PEO(4:1)SE的ASSLB中,LiCoO2上的CEI层很薄且均匀(图5h),而在含有PEO(16:1)SE的ASSLB中,LiCoO2的CEI层厚度约为3-6nm,且不均匀(图5i)。
因此,高浓度PEO SE可以抑制界面降解动力学,抑制正极电解质间相(CEI)中电子传导的产生,抑制LiCoO2到Co3O4的不可逆相变,从而形成薄而均匀的CEI层(图5j),这有助于提高Li/PEO/LiCoO2系统的高容量电化学性能。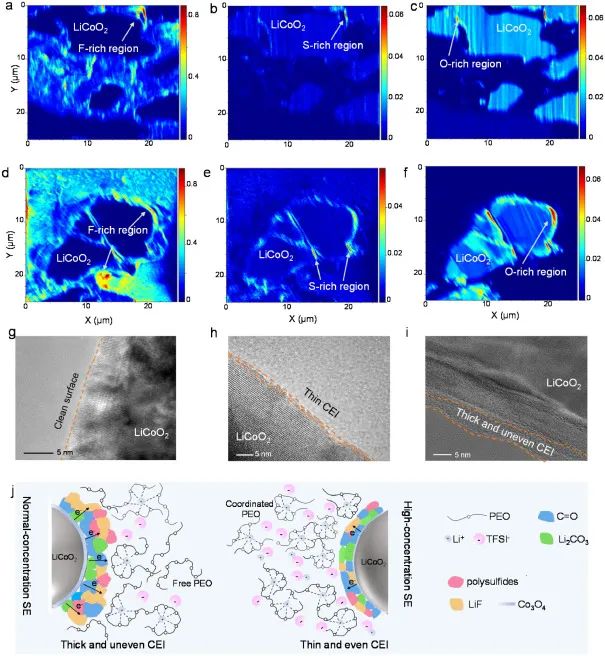
图5. CEI层的组成和结构。(a, d)F-, (b, e)S-和(c, f)O-段分布,通过TOF-SIMS获得,在使用(a-c)PEO(16:1)/PEO(4:1)和(d-f)PEO(16. 1)从电池中回收的复合正极。1)SE,以及(g)原始钴酸锂和使用(h)PEO(16:1)/PEO(4:1)和(i)PEO(16:1)SE的ASSLBs在20个循环后的高分辨TEM图像。(j) PEO SEs的配位结构和CEI层上的结果功能。
总结与展望
总之,高氧化电位的PEO SEs(> 5 V vs. Li/Li+)是基于浓盐化学设计的,它使大多数EO链参与配位结构,PEO(4:1)SE的氧化电位提高到约5.19 V。双层PEO(16:1)/PEO(4:1)SE与4.2V级别的LiCoO2和NMC(622)正极结合时,正极一侧的高浓度PEO(4:1)拥有明显改善的电化学性能。
高浓度的PEO SE减缓了界面降解动力学,减轻了CEI层的电子传导和LiCoO2的不可逆相变,从而实现了快速的界面电荷转移和整洁、薄而均匀的CEI层。该研究结果为安全和长程电动汽车提供高电压和高能量密度的锂电池阐明了PEO SEs的设计策略。
审核编辑:刘清
-
聚合物锂电池的生产2013-05-10 0
-
锂电池VS聚合物锂电池,谁才是未来的主角?2018-08-17 0
-
锂离子电池聚合物电解质导电机理是什么?2009-10-29 7146
-
锂离子电池聚合物电解质导电机理2009-12-09 2284
-
固态聚合物锂电池中电解质的技术研究2020-06-05 5706
-
中科院设计出一种用于柔性全固态锂金属电池的固体聚合物电解质2021-05-26 4048
-
原位固态化聚合物电解质基高性能准固态软包锂电池2022-06-22 8377
-
聚合物固态电解质的合理设计2022-08-18 1299
-
氟化石墨烯增强聚合物电解质用于固态锂金属电池2022-09-28 2755
-
钠离子电池的电解质分类2022-10-09 4882
-
高熵微区互锁的全固态聚合物电解质2022-11-10 2050
-
固态电池电解质的分类及性能对比2022-11-30 16316
-
聚合物电解质离子电导率及界面稳定性的影响因素2023-02-03 3432
-
聚苯并咪唑助力高性能富盐体系聚合物电解质锂离子电池2023-04-11 1436
-
请问聚合物电解质是如何进行离子传导的呢?2024-03-15 1236
全部0条评论

快来发表一下你的评论吧 !
